Modelling

Molekulardynamik (MD)
Unter Molekulardynamik, kurz MD, versteht man die computergestützte Berechnung molekularer Bewegungen und Interaktionen in atomarer Auflösung. Mit Hilfe sog. Kraftfelder, die Parameter für verschiedenste Moleküle bereitstellen (atomare Partialladungen, Bindungslängen, Bindungswinkel, Torsionswinkel, van der Waals- und Coulomb-Terme), kann eine detailgetreue Vorhersage einer Vielzahl molekularer Prozesse erreicht werden. Häufige Anwendungsgebiete sind u.a. Untersuchungen der dreidimensionalen Struktur eines Moleküls in Abhängigkeit verschiedener Lösungsmittel, die Aufklärung des Diffusionsverhaltens eines gelösten Stoffes, die Vorhersage von Wechselwirkungen zwischen Substanzen und biologischen Rezeptoren, die Bestimmung bindungsrelevanter Strukturparameter oder die Berechnung thermodynamischer und kinetischer Größen von Gleichgewichtssystemen.
In unserer Arbeitsgruppe wird die Molekulardynamik u.a. dafür eingesetzt, Orientierungen kleiner organischer Moleküle in partiell ausgerichteten Polymergelen vorherzusagen. Da die Messung dipolarer Restkopplungen (RDCs) durch NMR-Spektroskopie eine immer wichtigere Rolle bei der Bestimmung der räumlichen Struktur und der Wechselwirkungen biologisch aktiver Substanzen einnimmt, könnte eine exakte Berechnung der RDCs sowohl zur Überprüfung der Qualität gemessener NMR-Daten als auch direkt für die Strukturrechnung (im Falle fehlender experimenteller Daten) verwendet werden.
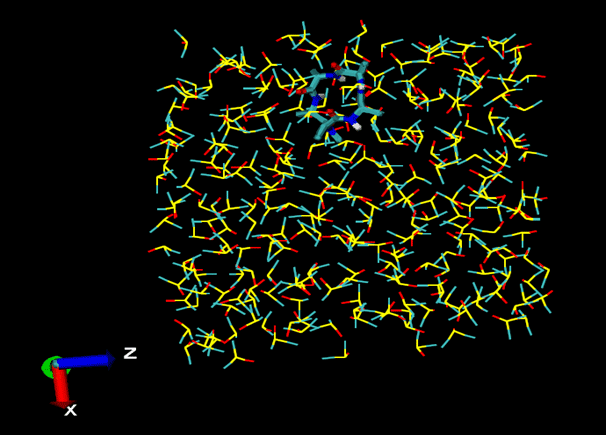
MD-"Simulationsbox": Cyclisches Peptid (Mitte oben) in einer DMSO-Solvensumgebung
Programme: GROMACS, GROMOS, SYBYL, INSIGHT
Virtuelles Protein-Protein- und Protein-Ligand-"Docking":
Biologische Systeme basieren auf der Wechselwirkung zwischen verschiedenen Biomolekülen (z.B. Proteine, Nukleinsäuren, kleine organische Moleküle) innerhalb einer Zelle. Diese Interaktionen sind essentiell für die Aufrechterhaltung des Stoffwechsels (und damit des Überlebens einer Zelle oder des gesamten Organismus) und der Anpassung an verschiedene Umweltbedingungen. Eine Störung dieses fein ausgewogenen Wechselwirkungsgleichgewichts kann verschiedene Krankheiten wie z.B. Krebs oder Diabetes hervorrufen. Aus diesem Grund ist die Untersuchung von molekularen Interaktionen eine überaus wichtige Disziplin innerhalb der biochemischen Grundlagenforschung und der medizinisch-pharmazeutischen Chemie. Neben biologischen Assays oder biophysikalischen Messmethoden, in denen Wechselwirkungspartner und Bindungsregionen identifiziert werden können, stellt das computergestützte "Docking" eine elegante Methode zur Vorhersage von potentiellen Interaktionen dar. Vorraussetzung für ein erfolgreiches "Docking" ist die Verfügbarkeit der dreidimensionalen Struktur der Wechselwirkungspartner (NMR-, Röntgenkristallographie- oder durch Homologie-Modellierung gewonnene Strukturen) sowie (nicht unbedingt notwendige) Informationen über mögliche Bindungsstellen und -modi. Die den "Docking"-Programmen zugrundeliegenden Algorithmen und damit die Qualität der Vorhersage unterscheiden sich teilweise stark, weswegen eine Kombination mehrerer Programme empfohlen wird. Neben dem sog. "ab initio Docking", bei dem keinerlei messtechnisch gewonnenen Informationen für das "Docking" verwendet werden, können - falls vorhanden - biophysikalische Messdaten den Programmen zur Verfügung gestellt werden, was zu wesentlich besseren und glaubwürdigen "Docking"-Resultaten führt.
Unser Interesse gilt einerseits der Aufklärung von Interaktionen zwischen Proteinen und natürlichen Liganden als auch der Vorhersage von Wechselwirkungen zwischen chemisch synthetisierten Substanzen und potentiellen Rezeptorproteinen. Das Hauptaugenmerk liegt dabei auf der Suche nach neuen, bioaktiven Integrin-Liganden und der Untersuchung von Wechselwirkungen zwischen Chaperonen, p53 und neu-entwickelten Liganden, was vor allem für das Verständnis der Krebsentstehung wichtige Informationen liefern kann.
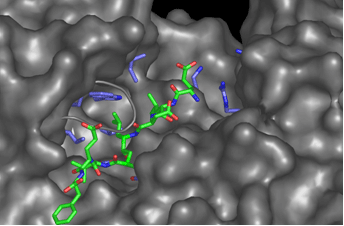
Docking eines Peptidmimetikums an das Integrin ανβ3
Programme: HADDOCK, FLEXX, AUTODOCK
Protein und Liganden basiertes in silico "Screening":
Während in früheren Jahren für die Suche nach neuen, bioaktiven Liganden für bestimmte Zielproteine ausschliesslich riesige Substanzbibliotheken in biologischen Essays "gescreent" wurden, bedient man sich heutzutage eines rationaleren in silico Ansatzes. Mit Hilfe des virtuellen "Screenings" kann man die Essay-basierte Suche nach aktiven Verbindungen stark vereinfachen, indem durch das computerbasierte Durchsuchen virtueller Ligandenbibliotheken bestimmte Strukturelemente identifiziert werden können, die für eine Wechselwirkung mit der Bindungstasche eines Rezeptorproteins essentiell sind. Wenn dreidimensionale Strukturdaten eines natürlichen Protein-Ligand-Komplexes zur Verfügung stehen, wird das "Screening" ebenfalls erleichtert, da hierdurch Informationen über die Bindungstasche und die an der Interaktion beteiligten Atome zugänglich sind. Zusätzlich können Messdaten aus Mutations- oder anderweitigen Bindungstudien verwendet werden, um die für eine Bindung verantwortlichen Atome (bzw. Aminosäurereste) zu charakterisieren. Für das Durchsuchen der Datenbanken werden sog. Pharmakophormodelle erstellt, die -. basierend auf den vorher beschriebenen Informationen - physikalische und geometrische Zwangsbedingugen darstellen. Solche virtuellen Liganden, die alle Bedingungen innerhalb festgelegter Toleranzen erfüllen, werden als potentielle Binder identifiziert und können anschließend sowohl mit Hilfe der "Docking"-Programme als als auch experimentell (nach erfolgreicher Synthese) auf ihr Bindungspotenzial hin überprüft werden.
Die rechnergestützte "Screening"-Technologie findet in unserem Arbeitskreis vielfältigen Einsatz. Als Zielmoleküle werden vor allem solche Proteine gewählt, die essentielle Funktionen innerhalb der Zelle verrichten und dadurch an der Entstehung schwerwiegender Erkrankungen beteiligt sind. Zu diesen gehören z.B. verschiedene Integrine, molekulare Chaperone oder Adaptorproteine bestimmter Tumorsuppressor-Proteine.
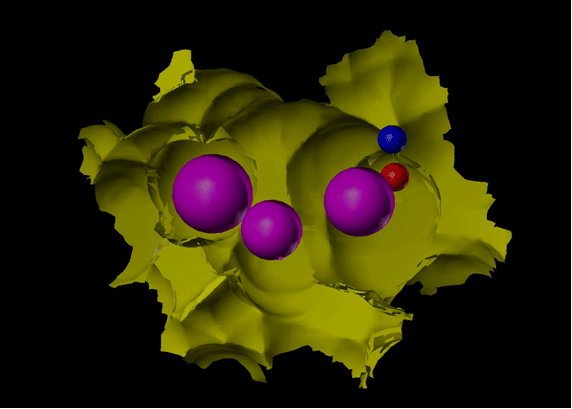
Pharmakophormodell für das "Screening" nach neuen Liganden für ein bestimmtes Zielprotein
Programm: UNITY
Strukturrechnung und Konformationsanalyse
Für das Verständnis biologischer Prozesse ist die Kenntnis der dreidimensionalen, atomaren Struktur zellulärer Moleküle unabdingbar. Gerade für weiterführende Studien wie die vorhergenannten "Docking"- oder "Screening"-Methoden stellt die Strukturrechnung alle wichtigen Basisinformationen zur Verfügung. Des Weiteren ist eine "fehlerfreie" Funktion von bioaktiven Molekülen (vor allem von Proteinen) stark von deren 3D-Struktur abhängig. Als Beispiel sei an dieser Stelle das zelluläre Prionprotein genannt, dessen Fehlfaltung nach heutigem Kenntnisstand die menschliche Creutzfeld-Jakob-Krankheit bedingt. Um dreidimensionale Molekülstrukturen ermitteln zu können, werden Abstands- (NOEs / ROEs) und Winkel- (skalare J-Kopplung) und Orientierungsinformationen (RDCs) aus NMR-Experimenten gewonnen und mit Hilfe der Berechnungsprogramme in geometrische Zwangsbedingungen "übersetzt". Eng mit der Strukturrechnung ist die Konformationsanalyse kleinerer Moleküle verbunden. Da molekulare Interaktionen auf dem sog. "Schlüssel-Schloss"-Prinzip beruhen, ist die Bestimmung der genauen Geometrie von flexiblen Molekülen eine wichtige Voraussetzung bei der Identifizierung natürlicher Liganden oder der Suche nach neuen, bioaktiven Substanzen. Hierbei muss berücksichtigt werden, dass die Konformation vor allem. von kleineren und flexiblen Molekülen wie z.B. (Cyclo-)Peptiden stark von der Lösungsmittelumgebung abhängt, was eine sorgfältige, rechnergestützte Analyse unter Berücksichtigung der Solvensumgebung bedingt. Durch den rasanten Anstieg der Computerleistungen können heutzutage die Strukturen selbst größerer Molekülsysteme in ihrer natürlichen Lösungsmittelumgebung berechnet werden.
In unserer Arbeitsgruppe werden sowohl die Strukturen großer Proteine und Proteinkomplexe aufgeklärt als auch das systematische Absuchen des Konformationsraumes kleinerer Systeme durchgeführt. Im Mittelpunkt des Interesses stehen hierbei bevorzugt Proteine, die wichtige Aufgaben während der zellulären Apoptose innehaben sowie verschiedene Chaperone. Des Weiteren gilt unser Interesse der systematischen Konformationsanalyse von cyclischen Peptiden, wobei der Fokus auf dem Einfluss verschiedender Aminosäure-Seitenketten und Manipulationen der Peptidbindung auf die dreidimensionale Konformation der Cyclopeptide liegt.
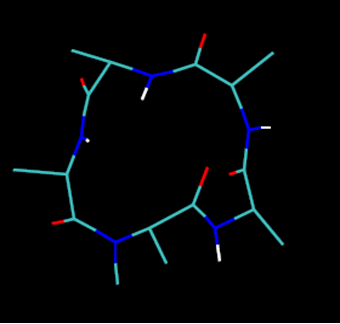
Strukturelle Konformation eines cyclischen Pentapeptids
Programme: DISGEO, INSIGHT, SYBYL, X-PLOR, GROMACS